Гидроочистка дизельных топлив
+
Алкилбензолы на катализаторах с высокой гидрирующей активностью подвергаются дальнейшему гидрогенолизу, в основном с последовательным отщеплением метана:
wspan=2 align=left valign=top> |  | ||||
 |
![]()
![]()
![]()
|
Механизм и кинетика процесса гидроочистки
Механизм гидрирования сераорганических соединений в значительной степени зависит от их строения. Скорость гидрирования, в общем, возрастает в ряду: тиофены < тиофаны » сульфиды < дисульфиды < меркаптаны.
Данных о гидрировании азот- и кислородорганических соединений очень мало. В таблице 4 приведены данные о гидрировании некоторых азот-, кислород- и сераорганических аналогов на Ni2S3 [4].
Таблица 4 – Степень превращения различных видов гетероатомных соединений в зависимости от температуры
| Углеводород |
Превращение, % | ||
| при 200°С | при 350°С | при 400°С | |
|
Тиофан | 41 | 100 | 100 |
|
Тетрагидрофуран | 0 | 25 | 55 |
|
Тиофен | 0 | 15 | 39 |
|
Фуран | 0 | 0 | 10 |
|
Пиррол | 0 | 0 | 0 |
При одинаковом строении устойчивость относительно гидрирования возрастает в ряду соединений: сераорганические < кислородорганические < < азоторганические.
Кинетика гидроочистки реальных промышленных видов сырья весьма сложна. Сложность определятся различием в скоростях превращения различных классов сернистых соединений (иногда на порядок больше), а также изменением активности катализатора в ходе процесса. Кроме того, всегда, особенно в случае тяжёлых продуктов, приходится считаться с большой вероятностью диффузионных ограничений. Наконец, влияют явления торможения реакций сероводородом при гидрогенолизе индивидуальных соединений. Несмотря на все перечисленные трудности, было выведено достаточно много кинетических уравнений для расчёта скоростей гидроочистки.
В одной из первых работ [5] было предложено уравнение первого порядка:
 ,
,
где ![]() и
и ![]() – парциальное давление сернистых соединений в гидрогенизате и в сырье,
– парциальное давление сернистых соединений в гидрогенизате и в сырье, ![]() – константа скорости реакции,
– константа скорости реакции, ![]() – условное время реагирования. Было показано, что до глубины обессеривания 95% и в случае узких фракций это уравнение удовлетворительно описывает скорость процесса. Однако для широких фракций оно не применимо, так как в этом случае скорость десульфуризации является суммой различных скоростей в уравнениях первого порядка для узких фракций. Константы скоростей десульфуризации, экстраполированные к нулевому парциальному давлению (бесконечное разбавление водородом), мало зависели от давления водорода, а соответствующие константы при парциальном давлении жидких продуктов 250 кПа – весьма существенно. Это интерпретировалось как явление более предпочтительной адсорбции жидких продуктов, вследствие чего при высоких парциальных давлениях последних поверхность катализатора становится труднодоступной для водорода и его давление начинает определять скорость реакции [5].
– условное время реагирования. Было показано, что до глубины обессеривания 95% и в случае узких фракций это уравнение удовлетворительно описывает скорость процесса. Однако для широких фракций оно не применимо, так как в этом случае скорость десульфуризации является суммой различных скоростей в уравнениях первого порядка для узких фракций. Константы скоростей десульфуризации, экстраполированные к нулевому парциальному давлению (бесконечное разбавление водородом), мало зависели от давления водорода, а соответствующие константы при парциальном давлении жидких продуктов 250 кПа – весьма существенно. Это интерпретировалось как явление более предпочтительной адсорбции жидких продуктов, вследствие чего при высоких парциальных давлениях последних поверхность катализатора становится труднодоступной для водорода и его давление начинает определять скорость реакции [5].
Позднее, вышеописанное уравнение было упрощено (не учитывалось влияние циркулирующего водорода):
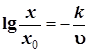 ,
,
где ![]() и
и ![]() – концентрация серы в сырье и продукте,
– концентрация серы в сырье и продукте, ![]() – объёмная скорость подачи сырья, а
– объёмная скорость подачи сырья, а ![]() – константа скорости реакции.
– константа скорости реакции.
Наконец, была показана применимость уравнения первого порядка, как по сырью, так и по водороду, выведенного на основании изотермы Ленгмюра. Однако приложение его к скоростям гидрогенолиза индивидуальных соединений показало столь значительную разницу, что уравнение пришлось сильно усложнить. Поэтому для промышленного сырья, особенно для сырья широкого фракционного состава или высококипящего, подбирали любые эмпирические уравнения, лишь бы они давали лучшую сходимость, чем уравнения первого порядка.
Так, на основании результатов опытов обессеривания вакуумного остатка кувейтской нефти с 5,45% серы при 3,5 и 7,0 МПа было выведено следующее уравнение [5]:
![]() ,
,
где ![]() – отношение содержания серы в продукте к содержанию её в сырье,
– отношение содержания серы в продукте к содержанию её в сырье,
![]() – константа скорости реакции;
– константа скорости реакции; ![]() – объёмная скорость. При этом авторы не считают, что второй кинетический порядок – истинный, просто он является лучшим приближением суммы многих уравнений первого порядка для отдельных классов и групп сернистых соединений. Вывод о кажущемся втором порядке подтверждён и в других работах.
– объёмная скорость. При этом авторы не считают, что второй кинетический порядок – истинный, просто он является лучшим приближением суммы многих уравнений первого порядка для отдельных классов и групп сернистых соединений. Вывод о кажущемся втором порядке подтверждён и в других работах.